Variante con pérdida de función en MAGT1 causante de la enfermedad de XMEN en un paciente colombiano con inmunodeficiencia común variable
Resumen
Introducción. La inmunodeficiencia común variable es un diagnóstico de exclusión en pacientes con mayor susceptibilidad a las infecciones, hipogammaglobulinemia, reacción deficiente a la vacunación o disminución de los porcentajes de linfocitos B de memoria en sangre. En los países de bajos y medianos ingresos, la definición y el estudio de los defectos moleculares en estos pacientes puede tardar décadas.
Objetivo. Dilucidar el defecto genético que produce la alteración de la inmunidad en un paciente con diagnóstico de inmunodeficiencia común variable.
Materiales y métodos. El fenotipo del paciente se obtuvo de la historia clínica. La expresión de NKG2D en células asesinas naturales se evaluó mediante citometría de flujo. Se realizó la secuenciación completa del exoma del paciente y de sus padres. Por medio de secuenciación por Sanger, se confirmó la variante patogénica.
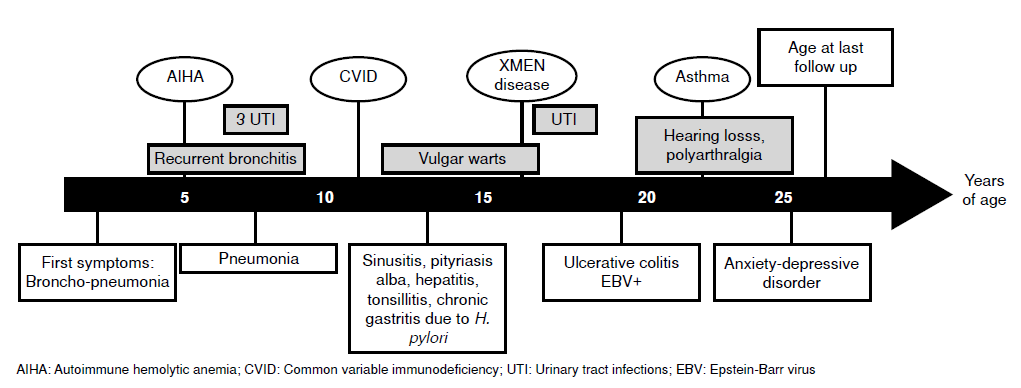
Resultados. El paciente reportó infecciones urinarias y de las vías aéreas superiores, además de anemia hemolítica autoinmunitaria y hepatopatía. Se observó una disminución de la expresión de NKG2D en las diferentes subpoblaciones sanguíneas de las células asesinas naturales. Los estudios serológicos y de carga viral para el virus de Epstein-Barr fueron positivos, pero, hasta la fecha, no se ha documentado neoplasia de las células B. El paciente presentó una mutación sin sentido en el exón 3 del gen MAGT1 (c.409C>T, rs387906724) en el cromosoma X, lo que resultó en una sustitución del aminoácido arginina por un codón de parada en la posición 137 de la proteína (R137X).
Descargas
Referencias bibliográficas
Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022;42:1473-507. https://doi.org/10.1007/s10875-022-01289-3
Peng XP, Caballero-Oteyza A, Grimbacher B. Common variable immunodeficiency: More pathways than roads to Rome. Annu Rev Pathol. 2023;18:283-310. https://doi.org/10.1146/annurev-pathmechdis-031521-024229
Poli A, Michel T, Thérésine M, Andrès E, Hentges F, Zimmer J. CD56 bright natural killer (NK) cells: An important NK cell subset. Immunol. 2009;126:458-65. https://doi.org/10.1111/j.1365-2567.2008.03027.x
Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999;93:190-7. https://doi.org/10.1006/clim.1999.4799
Schatorjé EJ, Gemen EF, Driessen GJ, Leuvenink J, van Hout RW, de Vries E. Paediatric reference values for the peripheral T cell compartment. Scand J Immunol. 2012;75:436-44. https://doi.org/10.1111/j.1365-3083.2012.02671.x
Morbach H, Eichhorn EM, Liese JG, Girschick HJ. Reference values for B cell subpopulations from infancy to adulthood. Clin Exp Immunol. 2010;162:271-9. https://doi.org/10.1111/j.1365-2249.2010.04206.x
Robbins JB, Austrian R, Lee CJ, Rastogi SC, Schiffman G, Henrichsen J, et al. Considerations for formulating the second-generation pneumococcal capsular polysaccharide vaccine with emphasis on the cross-reactive types within groups. J Infect Dis. 1983;148:1136-59. https://doi.org/10.1093/infdis/148.6.1136
García de Olarte D, Posada LH, García LF, Cardona R. Niveles de inmunoglobulinas séricas en población normal de Medellín. Acta Med Colomb. 1984;9:45.
Li FY, Chaigne-Delalande B, Kanellopoulou C, Davis JC, Matthews HF, Douek DC, et al. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature. 2011;475:471-6. https://doi.org/10.1038/nature10246
Chaigne-Delalande B, Li FY, O’Connor GM, Lukacs MJ, Jiang P, Zheng L, et al. Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D. Science. 2013;341:186-91. https://doi.org/10.1126/science.1240094
Ravell JC, Matsuda-Lennikov M, Chauvin SD, Zou J, Biancalana M, Deeb SJ, et al. Defective glycosylation and multisystem abnormalities characterize the primary immunodeficiency XMEN disease. J Clin Invest. 2020;130:507-22. https://doi.org/10.1172/JCI131116
Edgar D, Ehl S. The European Society for Immunodeficiencies (ESID) Registry - Working definitions for clinical diagnosis of PID. Accessed: October 9, 2024. Available at: https://esid.org/content/download/13053/372959/file/ESIDRegistry_ClinicalCriteriapdf
Brault J, Liu T, Bello E, Liu S, Sweeney CL, Meis RJ, et al. CRISPR-targeted MAGT1 insertion restores XMEN patient hematopoietic stem cells and lymphocytes. Blood. 2021;138:2768-80. https://doi.org/10.1182/blood.2021011192
Torgerson TR. Is this a cure for XMEN? Blood. 2021;138:2743-4. https://doi.org/10.1182/blood.2021012755
Derechos de autor 2024 Biomédica

Esta obra está bajo una licencia internacional Creative Commons Atribución 4.0.
| Estadísticas de artículo | |
|---|---|
| Vistas de resúmenes | |
| Vistas de PDF | |
| Descargas de PDF | |
| Vistas de HTML | |
| Otras vistas | |
Datos de los fondos
-
Ministerio de Ciencia, Tecnología e Innovación
Números de la subvención 1115-569-34430